文章来源:健康时报 2020-06-09 12:02
降低冠心病首次心脏病发作或卒中风险!FDA为阿斯利康替格瑞洛开绿灯
阿斯利康宣布,Brilinta(替格瑞洛)已在美国获批用于降低高危冠心病(CAD)患者的首次心脏病发作或卒中风险,CAD是最常见的心脏病类型。这是监管机构首次批准阿司匹林联合替格瑞洛,双重抗血小板治疗心血管(CV)高风险但无心脏病发作或卒中病史的患者。该药最早于2011年7月20日获FDA批准用于急性冠状动脉综合征(ACS)患者,降低血栓性心血管事件的发生率,2012年11月22日获中国药监局批准,用于在急性冠脉综合征(不稳定性心绞痛、非ST段抬高心肌梗死或ST段抬高心肌梗死)患者中,降低血栓性心血管事件的发生率。

美国FDA的批准是基于3期临床试验THEMIS的积极结果。该试验显示,在具有首次心脏病发作或卒中高危风险的冠心病患者和2型糖尿病(T2D)患者中,与单用阿司匹林相比,阿司匹林联合替格瑞洛治疗36个月时主要不良心血管事件的主要复合终点具有统计学显著性降低。主要复合终点是心脏病发作和卒中的减少。
THEMIS是一项多国家、随机双盲3期临床试验,旨在检验替格瑞洛和阿司匹林联合用药可减少主要不良心血管事件(MACE)风险的这一假设。THEMIS试验于2014年初启动,是目前为止在2型糖尿病患者中开展的最大规模随机试验之一。CAD定义为经皮冠状动脉介入治疗(PCI)、搭桥手术或冠状动脉至少50%狭窄。THEMIS试验表明,在无心脏病发作或卒中病史的CAD和T2D患者中,与阿司匹林单药治疗相比,阿司匹林联合长期替格瑞洛治疗使心脏病发作、卒中和CV死亡复合终点的相对风险降低10%(绝对风险降低;0.8%,7.7% vs 8.5%)。
“刺杀”特定肺癌!礼来VEGFR抑制剂组合获FDA批准
礼来(Lilly)公司宣布,美国FDA已经批准该公司的Cyramza(ramucirumab)与厄洛替尼(erlotinib)联用,一线治疗伴有表皮生长因子受体(EGFR)外显子19缺失或外显子21(L858R)突变的转移性非小细胞肺癌(NSCLC)患者。获得这一批准后,Cyramza现在已经获得了六个FDA批准,用于治疗某些类型的肺癌、肝癌、胃癌和结直肠癌。
Cyramza联合厄洛替尼是首个FDA批准,一线治疗转移性EGFR突变非小细胞肺癌的VEGFR/EGFR酪氨酸激酶抑制剂组合疗法。这一批准是基于全球性、随机、含安慰剂对照的3期临床试验RELAY的疗效和安全性数据。

在RELAY研究中,Cyramza(血管内皮生长因子受体2拮抗剂)与厄洛替尼(EGFR抑制剂)联用,为患者的无进展生存期提供具有统计学意义和临床意义的改善。Cyramza组合疗法组的PFS为19.4个月,对照组为12.4个月。(HR=0.59;CI,0.46,0.76;p<0.0001)。
RELAY是Cyramza治疗转移性非小细胞肺癌的第二个获得积极结果的3期临床试验。第一个是REVEL,它支持批准Cyramza联合多西他赛作为转移性非小细胞肺癌患者的治疗方案,这些患者的癌症在接受铂类化疗后继续进展。
随访时间长达4年!A型血友病基因疗法长期疗效喜人
BioMarin Pharmaceutical宣布了其治疗成人重度A型血友病的研究性基因疗法valocococogene roxaparvovec,在开放标签1/2期临床研究中的最新结果。长期随访结果表明,在接受一次基因疗法后4年时,患者仍然不需要接受其它预防性疗法。该数据已作为最新摘要提交给将于今年6月14—19日举行的世界血友病联盟(WFH)虚拟峰会。这款基因疗法的上市申请目前正在接受美国FDA和欧盟EMA的审评。

Valoctocogene roxaparvovec是一种使用AAV5病毒载体递送表达因子VIII的转基因的基因疗法。它的优势在于患者可能只需要接受一次治疗,肝细胞就可以持续表达因子VIII,从而避免需要长期接受预防性凝血因子注射。该疗法已经获得美国FDA授予的突破性疗法认定和欧盟授予的PRIME药品认定,以及EMA和FDA授予的孤儿药资格。然而,对这一基因疗法的疑虑在于它的疗效能够维持多久。最新的临床数据在这方面提供了更多信息。新闻稿指出,这是迄今为止,对血友病基因疗法最长时间的随访结果之一。
最新的试验结果表明,接受剂量为6e13 vg/kg的基因疗法治疗的患者和接受剂量为4e13 vg/kg基因疗法治疗的患者自接受valococogene roxaparvovec单次给药后仍未接受预防性凝血因子VIII的治疗。这些患者的随访时间分别达到了4年和3年。 在接受治疗后的第4年中,6e13 vg/kg队列的平均年出血率(ABR)为1.3,在接受基因疗法治疗的第3年中,4e13 vg/kg队列的平均ABR为0.5。在过去的一年中,6e13 vg/kg队列的7名参与者中的6名没有出现自发性出血事件。4e13 vg/kg队列的6名参与者中的5名仍然没有出现自发性出血事件。患者血液中的凝血因子VIII活性水平虽然有所下降,但下降幅度与最近几年的观察结果相当,并仍然保持在提供止血功效的范围内。
剑指中轴型脊柱关节炎!礼来IL-17A抑制剂斩获FDA第5项批准
礼来(Lilly)公司宣布,美国FDA已经批准了该公司开发的IL-17A拮抗剂Taltz(ixekizumab)扩展适应症,治疗有炎症客观症状的活动性非放射学中轴型脊柱关节炎(nr-axSpA)患者。新闻稿指出,这一批准使Taltz成为首个被FDA批准用于nr-axSpA的IL-17A拮抗剂。
Taltz于2016年3月首次获得FDA批准,用于治疗中度至重度斑块状银屑病。随后,它还获得批准治疗活动性银屑病关节炎(PsA)成人患者的治疗,以及强直性脊柱炎。

Taltz的安全性和有效性在一项多中心、随机双盲、含安慰剂对照的3期临床研究中得到了证实,该研究纳入了具有客观炎症症状的活动性nr-axSpA成人患者。研究的主要终点是第52周达到ASAS40的患者比例。试验结果表明,达到主要终点的Taltz患者比例优于安慰剂,每4周接受Taltz (80 mg)治疗的患者中有30%达到ASAS40应答,而接受安慰剂治疗的患者中有这一数值为13%(P=0.0045)。主要的次要终点是第16周的ASAS40应答,Taltz组有35%的患者达到该终点,而安慰剂组这一数值为19%(P<0.01)。
降低MM患者疾病进展风险近五成!赛诺菲CD38抗体组合表现抢眼
赛诺菲(Sanofi)公司宣布,其抗CD38抗体Sarclisa(isatuximab),与卡非佐米和地塞米松(Kd)标准治疗联用,在治疗复发性多发性骨髓瘤(MM)患者的3期临床试验中达到主要终点。与卡非佐米和地塞米松构成的标准治疗相比,Sarclisa组合使疾病进展或死亡风险降低47%(HR=0.531,p=0.0007,n=179)。Sarclisa组合与Kd单独治疗相比,在多个亚组中显示了一致的治疗获益。
Sarclisa是一种单克隆抗体,可与MM细胞上CD38受体上的特异性表位结合。其通过多种作用机制发挥作用,包括程序性肿瘤细胞死亡(凋亡)和免疫调节活性。CD38在MM细胞表面高度均匀表达,使其成为基于抗体的治疗药物(如Sarclisa)的潜在靶点。

这项名为IKEMA的随机、多中心、开放标签3期临床试验在16个国家的69个中心入组了302例复发性MM患者。截至中期分析时,接受Kd治疗组的中位无进展生存期(PFS)为19.15个月,而接受Sarclisa联合治疗的患者的中位PFS尚未达到。
同时,Sarclisa联合治疗组中的完全缓解率(CR)为39.7%,标准治疗组为27.6%。在Sarclisa联合治疗组中29.6%的患者与标准治疗组中13%的患者获得最小残余病灶(MRD)阴性完全缓解,表明近30%的接受Sarclisa联合治疗的患者通过下一代测序(NGS)手段检测不到MM的存在。
“抓捕”难治性神经母细胞瘤!创新GD2抗体获FDA优先审评资格
Y-mAbs Therapeutics宣布,该公司为GD2抗体naxitamab递交的生物制品许可申请(BLA)已被美国FDA接受并授予优先审评资格,适应症为复发/难治性高危神经母细胞瘤患者。FDA预计将在今年11月30日前做出回复。FDA还表示,目前不计划召开咨询委员会会议讨论该申请。
Naxitamab是一款靶向GD2抗原的人源化单克隆抗体。GD2抗原表达在神经外胚层生成的肿瘤表面,包括神经母细胞瘤、黑色素瘤和骨肉瘤等肿瘤。Naxitamab通过与肿瘤表面的GD2抗原结合,能够引发抗体媒介的细胞毒性反应并激活免疫系统中补体系统,从而达到杀伤肿瘤的效果。它已被FDA授予孤儿药资格,用于治疗神经母细胞瘤和骨肉瘤;以及突破性疗法认定,用于与GM-CSF联合治疗高风险神经母细胞瘤。
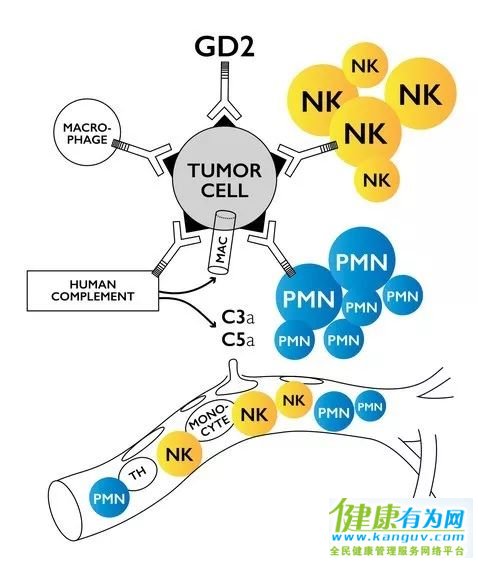
▲Naxitamab的作用机制(图片来源:Y-mAbs官网)
该申请的递交是基于一项名为12-230的1/2期临床试验的积极数据。在该试验的第一组神经母细胞瘤儿童患者中,28名不适合接受诱导化疗的复发/难治型高风险患者接受了naxitamab和GM-CSF的联合治疗,他们中有超过一半的患者也难以接受二线化疗的治疗。试验数据表明,该疗法使患者达到78%的客观缓解率(ORR),并使50%的患者无进展生存期(PFS)达到24个月。在第二组患者亚组中,30名接受挽救性疗法(salvage therapy)但效果不佳的患者接受了naxitamab和GM-CSF的联合治疗,其中有三分之一的患者的疾病曾复发过两次以上,并有89%的患者曾接受过抗GD2药物的治疗。试验数据表明,该疗法使患者达到37%的ORR,并使36%的患者PFS达到24个月。
完整3期临床结果发布!辉瑞口服JAK1抑制剂显著缓解皮炎症状
辉瑞(Pfizer)公司宣布,该公司开发的JAK1抑制剂abrocitinib治疗特应性皮炎的第二个关键性3期临床试验的完整结果在JAMA Dermatology上发布。Abrocitinib是一种研究性口服JAK1抑制剂(每日一次),在12岁及以上中重度特应性皮炎(AD)患者中使用。与第一项3期单药治疗研究一致,两种剂量的abrocitinib均达到所有共同主要终点和关键次要终点,且耐受性良好。
辉瑞的abrocitinib是一种口服小分子特异性JAK1抑制剂。JAK1抑制剂通过调节多种与特应性皮炎病理相关的细胞因子来控制病情,包括白细胞介素IL-4、IL-13、IL-31和干扰素γ。之前,abrocitinib已经获得美国FDA授予的突破性疗法认定。
JADE MONO-2是一项随机双盲、含安慰剂对照的平行组研究,旨在评价两种剂量(100 mg和200 mg,每日一次)abrocitinib单药治疗12周的疗效和安全性,共有391例中重度特应性皮炎受试者参与这一临床试验。
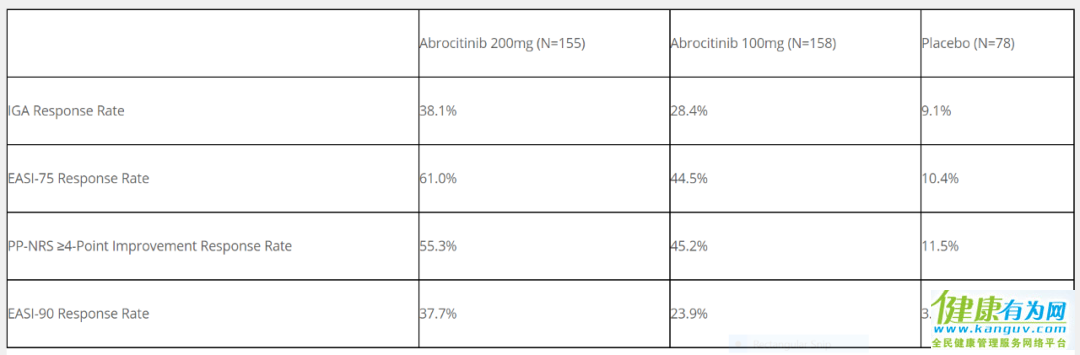
▲不同剂量的abrocitinib的疗效数据(图片来源:辉瑞公司官网)
试验结果表明,使用多种不同的症状检测方法,两种剂量的abrocitinib与安慰剂相比,都显著改善皮炎症状。
达到2期临床终点!渤健红斑狼疮创新单抗疗法显著改善皮肤症状
渤健(Biogen)在欧洲风湿病大会(EULAR 2020)上,公布了该公司开发的在研单抗疗法BIIB059治疗皮肤红斑狼疮(CLE)患者的2期临床数据。BIIB059是一种靶向血液树突状细胞抗原2(BDCA2)的全人源化IgG1单克隆抗体。试验结果表明,与安慰剂组相比,BIIB059显著改善了疾病症状。

BIIB059目前正在开发用于治疗CLE和系统性红斑狼疮患者。BDCA2是一种在人类免疫细胞浆细胞样树突细胞(pDC)上唯一表达的受体,它可以减少包括I型干扰素(IFN-I)在内的炎性细胞因子的产生,在狼疮的病理机制中起着重要的调节作用。
名为LILAC的随机双盲,含安慰剂对照的2期LILAC研究总计入组了264名参与者,包括活动性CLE和SLE患者。在治疗CLE患者的研究部分,132名患者接受了不同剂量的BIIB059的治疗。
试验结果表明,使用皮肤红斑狼疮疾病面积和严重程度指数活性(CLASI-A)评分,接受BIIB059治疗的患者在16周时皮肤症状获得剂量依赖性缓解,达到了试验的主要终点。接受50 mg、150 mg和450 mg BIIB059治疗的CLE患者的CLASI-A评分分别降低了38.8%(p=0.015)、47.9%(p<0.001)和42.5%(p=0.001),显著高于安慰剂组(14.5%)。




